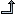Il sistema di farmacovigilanza in Italia
I motivi principali alla base della necessità di sorvegliare i farmaci anche dopo la loro introduzione in commercio sono rappresentati dai limiti delle sperimentazioni precliniche e cliniche.
La farmacovigilanza si pone come obiettivo generale quello di colmare le inevitabili lacune, sia in termini di attività terapeutica sia di tollerabilità, lasciate dalla sperimentazione pre-marketing e di verificare se il rapporto beneficio-rischio di un farmaco varia nel tempo, anche in relazione alle alternative terapeutiche disponibili.
L’evento che gettò le basi per la nascita di un sistema capillare e normato di FV fu la tragedia della talidomide che spinse la World Health Organization (WHO) a richiedere agli Stati membri, tra cui l’Italia, di istituire con urgenza reti nazionali di monitoraggio delle ADRs.
Nei primi anni ’60 Stati Uniti e Regno Unito avevano già istituito un sistema di raccolta dati e la yellow card sviluppata da Inman su richiesta del Dipartimento di salute britannico è stata presa come modello in tutti i Paesi e ancora oggi rappresenta il modo più semplice per riportare sospetti eventi avversi a farmaci.
Nel 1968 è stata avviata la centralizzazione dei dati mondiali sulle ADR, coordinata dalla World Health Organization (WHO) con il progetto “Program on International Drug Monitoring”, la cui banca dati mondiale è localizzata a Uppsala in Svezia (WHO-Uppsala Monitoring Centre for Pharmacovigilance, UMC: http://www.who-umc.org), a cui l’Italia aderisce nel 1975.
Il contesto normativo italiano inizia a delinearsi con la circolare n. 7 del 16 gennaio 1965, che invitava i medici ospedalieri a comunicare tempestivamente qualsiasi effetto tossico osservato durante il trattamento con farmaci, soprattutto se di recente introduzione; poi con l’art. 1 della legge del 7 agosto 1973, n. 519, che inseriva tra i compiti dell’Istituto Superiore di Sanità (ISS) l’accertamento della composizione e della innocuità dei prodotti farmaceutici di nuova istituzione prima della sperimentazione clinica sull’uomo; con la legge 883/1978 che favoriva lo sviluppo di “una disciplina della sperimentazione, produzione, immissione in commercio e distribuzione dei farmaci e dell’informazione scientifica sugli stessi, diretta ad assicurare l’efficacia terapeutica, la non nocività e la economicità del prodotto”.
Successivamente, con il DM del 1980 è stato previsto l’obbligo per le industrie farmaceutiche titolari di AIC di trasmettere al Ministero della Sanità un rapporto informativo periodico contenente «la specificazione della natura e della frequenza degli eventuali effetti tossici e secondari, sia locali che generali, conseguenti o comunque correlabili all’impiego di un farmaco, di cui i responsabili dell’impresa fossero venuti a conoscenza per diretta comunicazione della classe medica o per il tramite degli informatori scientifici o in qualunque altro modo» (art. 2). In caso di inadempimento come sanzione ci sarebbe stata la revoca dell’AIC.
Con il D.M. del 23 giugno 1981 l’obbligo a compilare le schede di segnalazione delle reazioni indesiderate connesse all’utilizzo di farmaci viene esteso anche ai medici.
Il termine “Farmacovigilanza” fa la sua comparsa nella legislazione italiana con la legge 531/1987 che stabiliva le disposizioni di farmacovigilanza a cui sottoporre i medicinali. La legge, inoltre, introdusse in Italia, prima che negli altri Paesi europei, la prassi della segnalazione spontanea da parte dei cittadini senza l’intermediazione degli operatori sanitari. Il sistema così normato determinò il bisogno di un organo che si occupasse della gestione e raccolta delle schede e del loro invio all’UMC. Per tali motivi, nel 1993 con il D.lgs. n. 266 venne istituita la Commissione Unica del Farmaco (CUF).
Contestualmente in Europa, con la Direttiva n.39 del 14 giugno e il Regolamento n.2309 del 22 luglio del 1993, si intervenne specificamente sulla fase successiva all’ottenimento dell’AIC stabilendo che «gli Stati membri istituissero un sistema di FV allo scopo di raccogliere informazioni utili per la sorveglianza dei medicinali, in particolare per quanto riguarda gli effetti collaterali negativi dei medicinali per l’uomo» (art. 29-bis); si rese inoltre obbligatoria la figura di un responsabile di FV per ciascuna società farmaceutica, così da assicurare la continuità del flusso informativo tra impresa e consumatori e redigere un rapporto dettagliato sui presunti effetti collaterali registrati, da inviare alle autorità competenti (art. 29-quinquies).
Si stabilì poi che gli Stati membri facessero pervenire «tutti i casi di presunti gravi effetti collaterali negativi» all’European Agency for the Evaluation of Medicines (EMEA), istituita a Londra nel 1995 (oggi European Medicine Agency- EMA), incaricata di coordinare la sorveglianza dei medicinali autorizzati e di garantire un uso sicuro ed efficacie degli stessi attraverso l’organizzazione e la libera consultazione di una banca dati di tutti gli effetti collaterali dei medicinali in questione.
La banca dati necessariamente informatizzata, che sarebbe stata alimentata dalle autorità competenti degli Stati membri, venne chiamata EudraVigilance (EV) e fu lanciata in via sperimentale nel 2001.
Intanto in Italia, il 18 febbraio 1997 venne emanata una nuova legge (DL n. 44 del 2/97), in attuazione della Direttiva 93/39 della CEE, che ribadiva l’obbligo per i medici di segnalare ogni presunta reazione avversa e lo estendeva anche ai farmacisti relativamente e solamente ai medicinali SOP (senza obbligo di prescrizione) e OTC (prodotti da banco).
Alla fine del 2001 il Ministero della Salute rese operativa la Rete telematica nazionale di farmacovigilanza (RNF) prevista dal DM del febbraio 1997, ma fino ad allora non attuata. Una nuova legge sulla farmacovigilanza venne emanata nell’aprile 2003 (D.lgs. n. 95 dell’8 aprile 2003), introducendo importanti modifiche ribadite con il D.lgs. 24 aprile 2006 n. 219:
- l’obbligo delle segnalazioni delle reazioni avverse da farmaci diventa esclusivamente di tipo deontologico; - si stabilisce attraverso specifiche linee guida cosa segnalare e la possibilità di segnalazione viene estesa a tutti gli operatori sanitari;
- i medici, i farmacisti e gli altri operatori sanitari sono tenuti a segnalare le ADR all’Azienda Sanitaria Locale (ASL) territorialmente competente o alla Azienda ospedaliera. Le ASL devono inserire le segnalazioni nella rete telematica nazionale (Rete nazionale di farmacovigilanza);
- l’ufficio di farmacovigilanza dell’AIFA, le aziende farmaceutiche interessate, le Regioni e i Centri Regionali ricevono dalla rete un avviso di questo inserimento. Le segnalazioni vengono poi inviate automaticamente all’Agenzia Europea dei Medicinali (EMA) e all’OMS.
I Centri Regionali di Farmacovigilanza (CRFV) sono stati inseriti nel Sistema italiano della segnalazione spontanea dal decreto legge del 2003 (D.L. 95/03) e sono diventati operativi all’interno della Rete nazionale di farmacovigilanza nel corso del 2006.
Nel 2010, Consiglio e Parlamento Europeo hanno deciso di affinare ulteriormente la regolamentazione della FV attraverso l’emanazione della Direttiva n. 2010/84 e del Regolamento n. 1235/2010.
Le modifiche più rilevanti introdotte dalla nuova legislazione sono:
- l’ampliamento del concetto di “reazione avversa” che include qualsiasi reazione nociva e non voluta conseguente non solo all’uso autorizzato di un medicinale secondo i termini dell’AIC ma anche agli errori terapeutici, all’uso improprio, all’abuso e agli usi non conformi;
- la visione del cittadino come soggetto attivo della FV con la facoltà di effettuare la segnalazione di ADR senza l’intermediazione dei medici (facoltà che in Italia esisteva già a partire dal D.L. 443 del 1987);
- il rafforzamento delle procedure di registrazione e comunicazione delle segnalazioni di ADRs e l’elaborazione di una procedura d’urgenza per il caso di dubbio fondato circa la sicurezza;
- l’istituzione del Comitato di valutazione dei rischi per la farmacovigilanza (PRAC), interno ad EMA, con la funzione di elaborare i pareri relativi alla gestione dei rischi derivanti dall’uso dei medicinali.
Sia la Direttiva n. 2010/84 che il Regolamento n. 1235/2010 sono divenuti operativi nel luglio 2012 e recepiti in Italia attraverso il DM 30 aprile 2015.
Punti chiave di partecipazione al sistema di FV delle figure coinvolte.
All’interno di questo sistema, secondo la normativa in vigore, i segnalatori devono inviare le segnalazioni ai Responsabili Locali di Farmacovigilanza (RAF o RLF) della propria struttura di appartenenza o alle aziende titolari di AIC.
I RLF devono inserire le segnalazioni ricevute in RNF entro sette giorni dalla data di ricevimento; verificarne la completezza e la congruità dei dati; archiviare le schede ed inviarne copia al Centro Regionale; eseguire follow-up dei casi ad esito fatale e di quelli gravi che necessitino di approfondimento; rispondere a eventuali richieste delle aziende farmaceutiche; fornire feedback ai segnalatori; diffondere informazioni agli operatori sanitari; essere un contact-point per AIFA su questioni di farmacovigilanza.
I titolari di AIC non sono più tenuti ad inviare le segnalazioni di sospette ADR alle autorità nazionali competenti (l’AIFA in Italia), ma devono trasmetterle direttamente al database Europeo di Farmacovigilanza “Eudravigilance” che, saranno poi inviate al database del WHO Uppsala Monitoring Centre.
I Centri Regionali di Farmacovigilanza devono svolgere un ruolo chiave, trovandosi in una posizione centrale tra l’Autorità Regolatoria da un lato e i Responsabili Locali dall’altro.
I CRFV attualmente gestiscono tutti gli aspetti di farmacovigilanza a livello locale e collaborano con l’AIFA per l’individuazione dei segnali d’allarme che possono emergere dall’analisi del database Eudravigilance.
Compiti e requisiti dei CRFV sono stati ben definiti a partire dall’Accordo Stato-regioni del 28/10/2010 e precisamente articolati e dettagliati con la POS AIFA approvata a giugno 2018.
Tra i compiti essenziali affidati ai CRFV vi sono la formazione e sensibilizzazione degli operatori sanitari; l’informazione e la divulgazione di argomenti di farmacovigilanza; il supporto ai RLFV nella raccolta ed inserimento delle segnalazioni di sospetta ADR nella RNF; controllo di codifica e qualità dei dati relativi alle segnalazioni della propria regione inserite nella RNF; valutazione del causality assessment; verifica e coordinamento delle attività di follow-up; analisi dei dati in collaborazione con AIFA.